




インフォマティクス初心者でも1細胞全ゲノムDNA解析が可能に
2020年11月24日
細胞は、増える。細胞は増えて、個体をかたちづくる。細胞が増える時、遺伝情報は細胞分裂の前にコピーされて2倍になる。コピーされるゲノムは、ヒトではなんと2 m、その中に60億塩基対が含まれる。この大量の情報をほぼ100%正確にコピーする仕組みを「ゲノムDNA複製」という。細胞の中で行われるこの非常にダイナミック、かつ繊細で複雑な生命現象を高解像度で解析するには、1細胞全ゲノム解析がお勧めだ。この解析方法を使えば、DNA複製異常も、さらには染色体異常も検出できる。1細胞全ゲノム解析は、やったことがないからできない?そう感じたら、これを読んでぜひ挑戦してみてほしい。難しそうだと諦める必要は、もうない。
理研BDRの三浦尚研究員、高橋沙央里研究員(発生エピジェネティクス研究チーム、平谷伊智朗チームリーダー)と三重大学の共同研究グループ*1は、今回、独自の1細胞全ゲノムDNA複製解析法「scRepli-seq」の実験と解析に必要な全てのノウハウを論文にまとめて公開した。本成果は科学誌 Nature Protocolsに2020年11月24日付で掲載された。
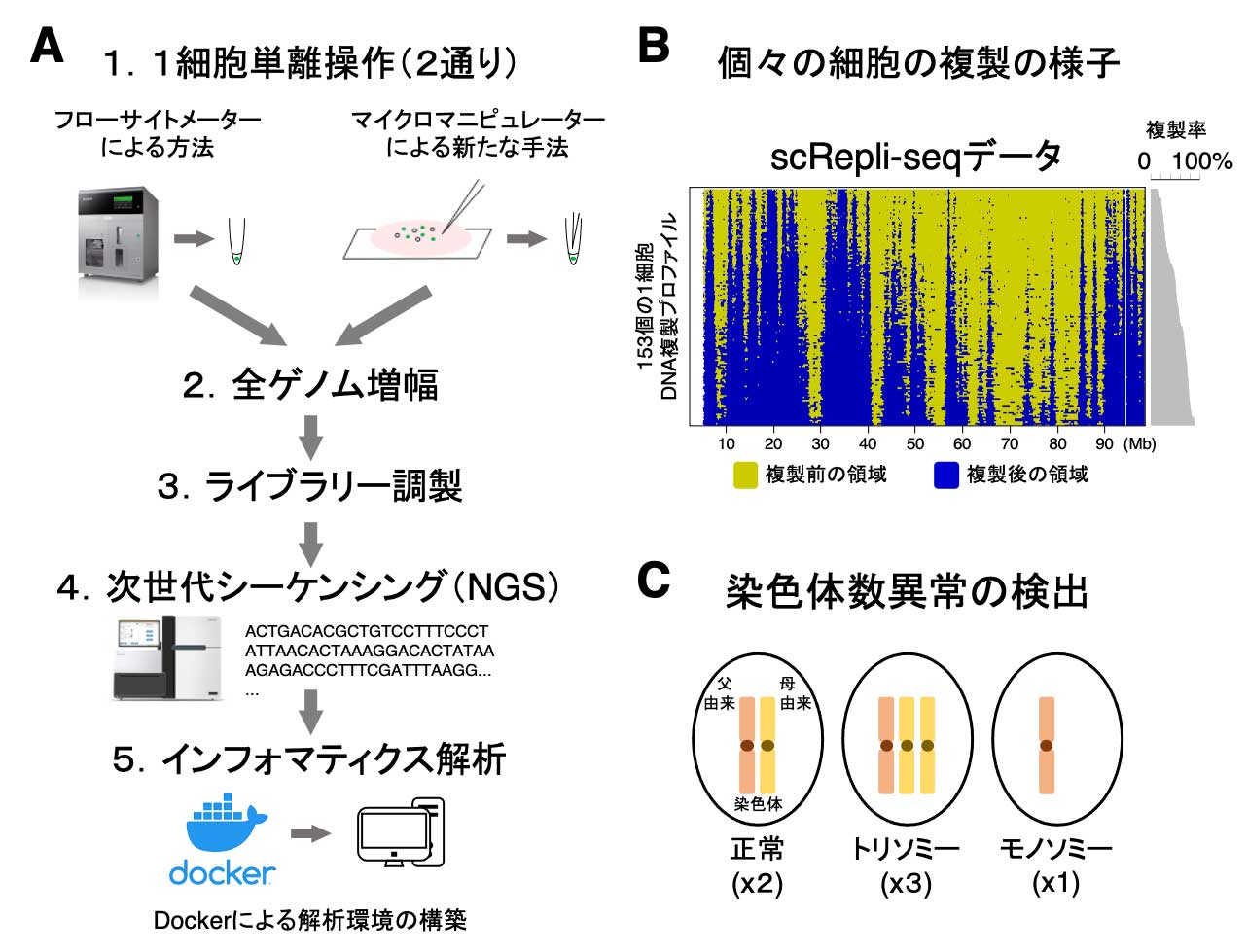
図.scRepli-seq法の概要と有用性
(A) scRepli-seq法の概要。(1) 1細胞単離操作では、フローサイトメーターを用いた標準的な方法とマイクロマニピュレーターを用いた新たな方法(希少細胞などに応用が可能)を示す。
(B) scRepli-seq法による1細胞レベルの全ゲノムDNA複製解析。細胞周期のS期全域から回収した153個の細胞の複製プロファイル。ゲノムDNAの複製率を算出し、ほとんど未複製な細胞から複製完了に近いものまでを縦に並べた。
(C) scRepli-seq法で観察可能な染色体数異常の例。トリソミーとモノソミーの模式図を示す。
酵母から哺乳類まで、ゲノムDNA複製の研究は、主に万単位の細胞集団を用いて展開されてきた。これは、1細胞レベルでゲノムDNA複製の真の姿を捉えることが難しかったからだが、最近、共同研究グループは、1細胞全ゲノムDNA複製解析法「scRepli-seq」を開発した*2(図. (A))。本解析法では、1個の細胞から取り出した全ゲノムDNAを均等に増幅して次世代シークエンサー(NGS)によって解読し、各領域のリード数の相対的な比から複製される前(1コピー)と複製された後(2コピー)の領域を識別することができる(図. (B))。このシンプルな原理で、ゲノムDNAが複製されていく様子が1細胞レベルで解析できるようになった。ただ、scRepli-seq法はDNA複製研究の枠にとどまらず、ゲノムDNA複製と密接にリンクしているクロマチン高次構造の変化も推定できる*3。さらに、染色体上の変異や染色体異常の検出にも利用できる。例えば、細胞におけるトリソミーやモノソミーの検出が可能で、ハプロタイプごとに分けた解析と組み合わせれば、染色体異常の由来が父母どちらのものであるかも判定できる(図. (C))。しかし、scRepli-seq法は実験とインフォマティクス解析の知識が要求されるため、初心者にはかなりハードルが高い。今回、三浦らは、scRepli-seq法を多くの研究者に広く使ってもらうために、実験と解析のプロトコルの詳細を余すところなく公開し、さらに未公開の方法論についても解説することで、この問題を解決しようと試みた。
まず、scRepli-seq法の実験部分については、エタノール固定した細胞標本を用いる標準的な方法を詳細に解説した。プロトコルは、大きく分けて、(1)細胞集団から1細胞を回収する(1細胞単離操作)、(2)細胞からゲノムDNAを抽出して増幅する(全ゲノム増幅)、(3)次世代シーケンシング(NGS)のためのライブラリーを調製する、という3つのステップからなる(図. (A))。この論文では、それぞれのステップについて、背景にある原理と陥りやすい罠などについて丁寧に解説した。また、105~106個の細胞を要するフローサイトメーターを用いる方法に対して、生体由来の希少細胞などへの応用としてマイクロマニピュレーターを用いる方法を新たに公開した(図. (A))。さらに、NGS解析前のサンプルの品質の判定基準も解説した。これにより、NGS解析前に品質の悪いサンプルを除外することで、品質の高いサンプルのみをNGS解析に回してシーケンシングの成功率を高めることが可能だ。
次に、scRepli-seq法の解析部分については、インフォマティクス初心者がつまずくことのないよう、必要な解析モジュールのインストールのステップから丁寧に解説した。実は、このインストールのステップはなかなかの曲者で、個々の解析モジュールのバージョンが違うだけで、一連の解析に不具合が生じてエラーが出ることが多く、無駄な時間を取られて挫折の要因になる。そこで、新たな解析環境「Docker」を使った解析スクリプトを作成し、紹介した(図. (A))。また、得られたNGSデータを評価する客観的基準についても解説した。
今後の課題は、解像度の向上だ。scRepli-seq法の解像度はまだ40 kb程度に留まる。これは平均約20 kbある遺伝子の倍近くにもなる。最近の細胞集団における全ゲノム解析では、50 bp程度のコピー数変化までが検出可能*4となり、個人間の多様な1–10 kb程度のコピー数多型が同定されている*5。また、様々ながん組織を用いた同様の解析により、がん細胞における新たなコピー数変化も見つかっている*6。特に、がん細胞集団内で、コピー数変化が細胞間でどのようにばらつき、それが、がんの悪性度とどのように関連しているのかはまだ不明な点が多いが、scRepli-seq法の解像度が向上すれば、それを詳しく解析できるだろう。また、解像度の向上とは別に、他の1細胞解析法と組み合わせた1細胞マルチオミックス解析も重要な課題であるが、「これらの課題は、十分に達成可能な目標である」と平谷チームリーダーは語る。
「scRepli-seq法のあらゆるステップについて、全くの初心者でも作業を確実に進められるよう、平易な解説を心がけたので、全ゲノム解析法にありがちな心理的なハードルはかなり下げられたと自負しています。」と話すのは三浦研究員。「本プロトコル論文をまとめていく過程で解析面での新たな発見がたくさんあり、評価基準が増えて、結果的に技術自体もさらに向上したと感じています。DNA複製にとどまらず、ゲノムDNAを扱うあらゆる研究者に本技術を使って頂ければ嬉しいです。」
高橋 涼香(BDR・広報グループ)
- *1: 本研究は以下の研究グループの共同研究で実施されました。
- 三浦 尚(理化学研究所 生命機能科学研究センター・研究員)・筆頭著者
- 高橋 沙央里(理化学研究所 生命機能科学研究センター・研究員)・共同筆頭著者
- 柴田 隆豊(三重大学大学院生物資源学研究科・修士課程学生(研究当時))・共同筆頭著者
- 長尾 恒治(大阪大学大学院理学研究科・准教授)
- 小布施 力史(大阪大学大学院理学研究科・教授)
- 奥村 克純(三重大学大学院生物資源学研究科・教授)
- 緒方 正人(三重大学大学院医学系研究科・教授)
- 平谷 伊智朗(理化学研究所 生命機能科学研究センター・チームリーダー)・共同責任著者
- 竹林 慎一郎(三重大学大学院生物資源学研究科・准教授)・責任著者
関連リンク
掲載された論文
関連記事
参考文献
*4: Altshuler DL, et al. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073(2010) doi: 10.1038/nature09534
*5: MacDonald J, et al. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 42: D986–D992(2014) doi: 10.1093/nar/gkt958
*6: ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578: 82-93(2020) doi:10.1038/s41586-020-1969-6