




当グループは、標的とする生体分子を制御するための分子を大規模計算機シミュレーションによって設計することを目標としています。近年X 線構造解析やNMR により、生体高分子の立体構造が次々と明らかになっています。また計測技術の発達によって、細胞内での分子の挙動も次第に観察できるようになってきました。しかし、立体構造が分子機能にどうつながるのかを予測するためには、原子スケールのモデルをもとにした長時間シミュレーションが必要になります。分子動力学計算や量子化学計算による分子シミュレーションを行うことで、生体高分子の機能の理解につなげます。センター内外との共同研究により、さまざまな標的分子に対して、培った手法を応用して新規化合物を設計します。また、分子動力学専用計算機を開発し、市販の計算機では不可能な長時間計算の実現を目指しています。
研究テーマ
- 生体高分子と制御分子および水和水の相互作用の、熱力学的、物理化学的特性の理解
- タンパク質/DNAと制御分子複合体の電子状態計算による触媒反応の解明
- 標的タンパク質に結合する新規ペプチド分子の設計
- 分子動力学専用計算機MDGRAPE-4A の開発
主要論文
Komatsu TS, Okimoto N, Koyama YM, et al.
Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation.
Scientific Reports
10, 16986 (2020)
doi: 10.1038/s41598-020-74099-5
Takaoka Y, Iwahashi M, Chini A, et al.
A rationally designed JAZ subtype-selective agonist of jasmonate perception.
Nature Communications
9, 3654 (2018)
doi: 10.1038/s41467-018-06135-y
Otsuka T, Okimoto N, Taiji M.
Assessment and acceleration of binding energy calculations for protein-ligand complexes by the fragment molecular orbital method.
Journal of Computational Chemistry
36(30), 2209-2218 (2015)
doi: 10.1002/jcc.24055
Yamagishi J, Okimoto N, Morimoto G, Taiji M.
A New Set of Atomic Radii for Accurate Estimation of Solvation Free Energy by Poisson-Boltzmann Solvent Model.
Journal of Computational Chemistry
35(29), 2132-2139 (2014)
doi: 10.1002/jcc.23728
Ohno Y, Yokota R, Koyama H, et al.
Petascale molecular dynamics simulation using the fast multipole method on K computer.
Computer Physics Communications
185(10), 2575-2585 (2014)
doi: 10.1016/j.cpc.2014.06.004
Ohmura I, Morimoto G, Ohno Y, et al.
MDGRAPE-4: a special-purpose computer system formolecular dynamics simulations.
Philosophical Transactions of the Royal Society a-Mathematical Physical and Engineering Sciences
372(2021), 20130387 (2014)
doi: 10.1098/rsta.2013.0387
Kondo HX, Taiji M.
Enhanced exchange algorithm without detailed balance condition for replica exchange method.
Journal of Chemical Physics
138(24), 244113 (2013)
doi: 10.1063/1.4811711
Kondo HX, Okimoto N, Morimoto G, Taiji M.
Free-Energy Landscapes of Protein Domain Movements upon Ligand Binding.
Journal of Physical Chemistry B
115(23), 7629-7636 (2011)
doi: 10.1021/jp111902t
Okimoto N, Futatsugi N, Fuji H, et al.
High-Performance Drug Discovery: Computational Screening by Combining Docking and Molecular Dynamics Simulations.
Plos Computational Biology
5(10), e1000528 (2009)
doi: 10.1371/journal.pcbi.1000528
ニュース

2023年3月2日 BDRニュース
BDRの研究ネホリハホリ
オリジナルマシンで「世界」と戦う
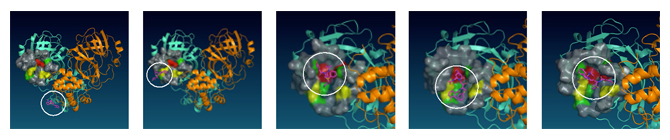
2020年11月10日 研究成果
新型コロナウイルスタンパク質の柔らかい構造
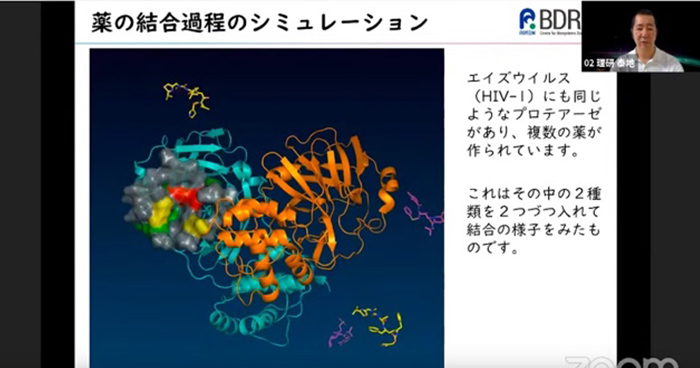
2020年8月21日 BDRニュース
一般向けイベント
中高生のための夏の特別授業「新型コロナウイルスに挑む生命科学・計算科学」を開催

2020年7月6日 研究成果
研究最前線:専用計算機が創薬の新時代を拓く(泰地真弘人チームリーダー)[PDF 2.1MB]

2020年4月24日 BDRニュース
小松輝久 研究員(計算分子設計研究チーム)のエッセイが産経新聞の連載「科学の中身」に掲載されました
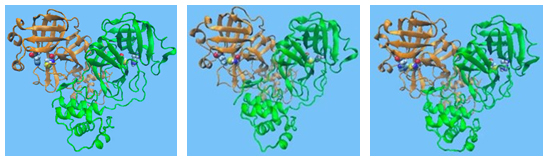
2020年3月23日 研究成果
新型コロナウイルス(SARS-CoV-2)メインプロテアーゼの分子動力学シミュレーションデータを公開

2019年11月18日 研究成果
創薬専用スパコンの開発
2018年9月18日 研究成果
植物の病原菌感染を防ぐ画期的な植物免疫強化剤を開発
