



Team Leader
Yuji Sugita
D.Sci.
Laboratory for Biomolecular Function Simulation
[Concurrent appointment to BDR ended in March 2025. He continues his research at RIKEN Pioneering Research Institute. ]
E-mailsugita@riken.jp
In our laboratory, we carry out molecular dynamics simulations on Fugaku or other supercomputers to elucidate the relationships between structure, dynamics, and function in biomacromolecules, such as proteins, nucleic acids, glycans, and lipid molecules. We develop various free-energy calculation methods to evaluate free-energy differences between two thermodynamic states in biomacromolecules for understanding biomolecular functions more quantitatively. The developed methods are installed into the GENESIS software, which is a freeware available to everyone. We thus contribute not only to the basic life science but also to in-silico drug discovery.
Research Theme
- Effects of intracellular crowding on the dynamics of protein and water interactions
- Simulations and data analysis of large-scale protein structural changes
- Slow dynamics using Brownian dynamics simulations
Selected Publications
Chong SH, Oshima H, Sugita Y.
Allosteric changes in the conformational landscape of Src kinase upon substrate binding.
Journal of Molecular Biology
(2024)
doi: 10.1016/j.jmb.2024.168871
Jung J, Yagi K, Tan C, et al.
GENESIS 2.1: High-Performance Molecular Dynamics Software for Enhanced Sampling and Free-Energy Calculations for Atomistic, Coarse-Grained, and Quantum Mechanics/Molecular Mechanics Models.
Journal of Physical Chemistry. B
128(25), 6028-6048 (2024)
doi: 10.1021/acs.jpcb.4c02096
Chyży P, Kulik M, Shinobu A, et al.
Molecular dynamics in multidimensional space explains how mutations affect the association path of neomycin to a riboswitch.
Proceedings of the National Academy of Sciences of the United States of America
121(15), e2317197121 (2024)
doi: 10.1073/pnas.2317197121
Shinobu A, Re S, Sugita Y.
Practical Protocols for Efficient Sampling of Kinase-Inhibitor Binding Pathways Using Two-Dimensional Replica-Exchange Molecular Dynamics.
Frontiers in Molecular Biosciences
9, 878830 (2022)
doi: 10.3389/fmolb.2022.878830
Matsubara D, Kasahara K, Dokainish HM, et al.
Modified Protein-Water Interactions in CHARMM36m for Thermodynamics and Kinetics of Proteins in Dilute and Crowded Solutions.
Molecules
27(17), 5726 (2022)
doi: 10.3390/molecules27175726
Oshima H, Sugita Y.
Modified Hamiltonian in FEP Calculations for Reducing the Computational Cost of Electrostatic Interactions
Journal of Chemical Information and Modeling
62(11), 2846-2856 (2022)
doi: 10.1021/acs.jcim.1c01532
Fujiyama K, Kato N, Re S, et al.
Molecular Basis for Two Stereoselective Diels-Alderases that Produce Decalin Skeletons*.
Angewandte Chemie
60(41), 22401-22410 (2021)
doi: 10.1002/anie.202106186
Kasahara K, Re S, Nawrocki G, et al.
Reduced efficacy of a Src kinase inhibitor in crowded protein solution.
Nature Communications
12(1), 4099 (2021)
doi: 10.1038/s41467-021-24349-5
Shinobu A, Kobayashi C, Matsunaga Y, Sugita Y.
Coarse-Grained Modeling of Multiple Pathways in Conformational Transitions of Multi-Domain Proteins.
Journal of Chemical Information and Modeling
61(5), 2427-2443 (2021)
doi: 10.1021/acs.jcim.1c00286
Re S, Oshima H, Kasahara K, et al.
Encounter Complexes and Hidden Poses of Kinase-Inhibitor Binding on the Free-Energy Landscape.
Proceedings of the National Academy of Sciences of the United States of America
116, 18404-18409 (2019)
doi: 10.1073/pnas.1904707116
Members
News
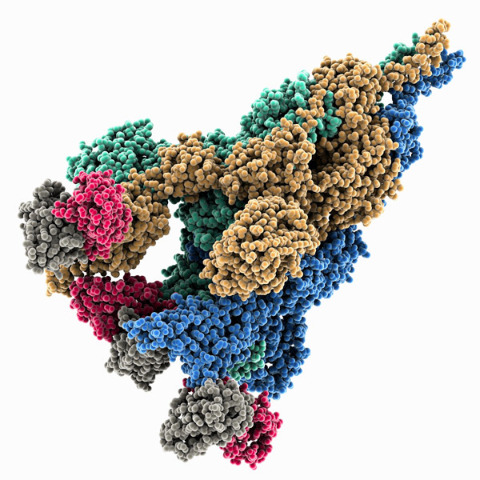
Mar. 27, 2023 Research
How an early mutation in the COVID-19 virus helped it spread so fast

Apr. 28, 2022 BDR News
Four BDR scientists awarded MEXT prizes
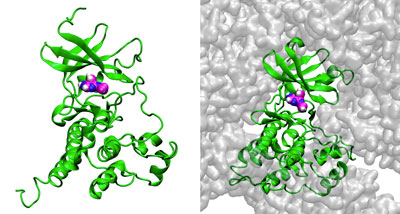
Oct. 21, 2021 Research
Supercomputer simulations reveal how protein crowding in cells impacts interactions

Jul. 30, 2021 BDR News
RIKEN BDR signs MOU with Shinshu University for research and educational collaborations
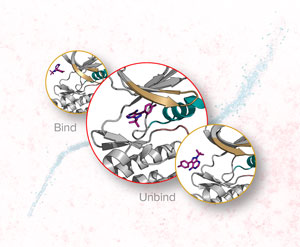
Nov. 29, 2019 Research
Faster modeling of interactions between ligands and proteins