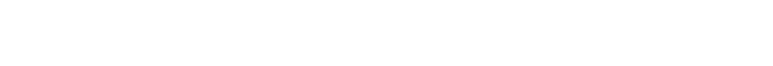




A Special-Purpose Computer for Molecular Dynamics SimulationsMDGRAPE-4A
We have been developing a series of special purpose computers for molecular dynamics simulations. In 2019 we have completed the MDGRAPE-4A, an improved version of MDGRAPE-4[1]. The target performance is to make it possible to simulate typical protein-ligand complex surrounded by water for microseconds per day, which is one order of magnitude faster than commercially available systems. Currently the full hardware system is in operation and production runs are on going on the system. Accuracy of the software was verified by measuring relative errors and comparing simulated physical properties with results of GROMACS single precision. The achieved performance is roughly 1 microsecond per day for the systems including about 100 thousand atoms. Utilizing MDGAPE-4A, a drug discovery project for COVID-19 is ongoing[2].
MDGRAPE-4A system architecture
MDGRAPE-4A SoC LSI
Advanced from MDGRAPE-4
Message flow (green arrows)
Data flow (blue arrows)
Design: RIKEN and D-Clue Technologies
Manufacturer: ALCHIP TSMC 40G 16.28mm x 16.28mm
Logic: ~50M gates
SRAM: ~72M bits
Power: 85W
Clock: 0.6GHz, 0.8GHz (pipelines)
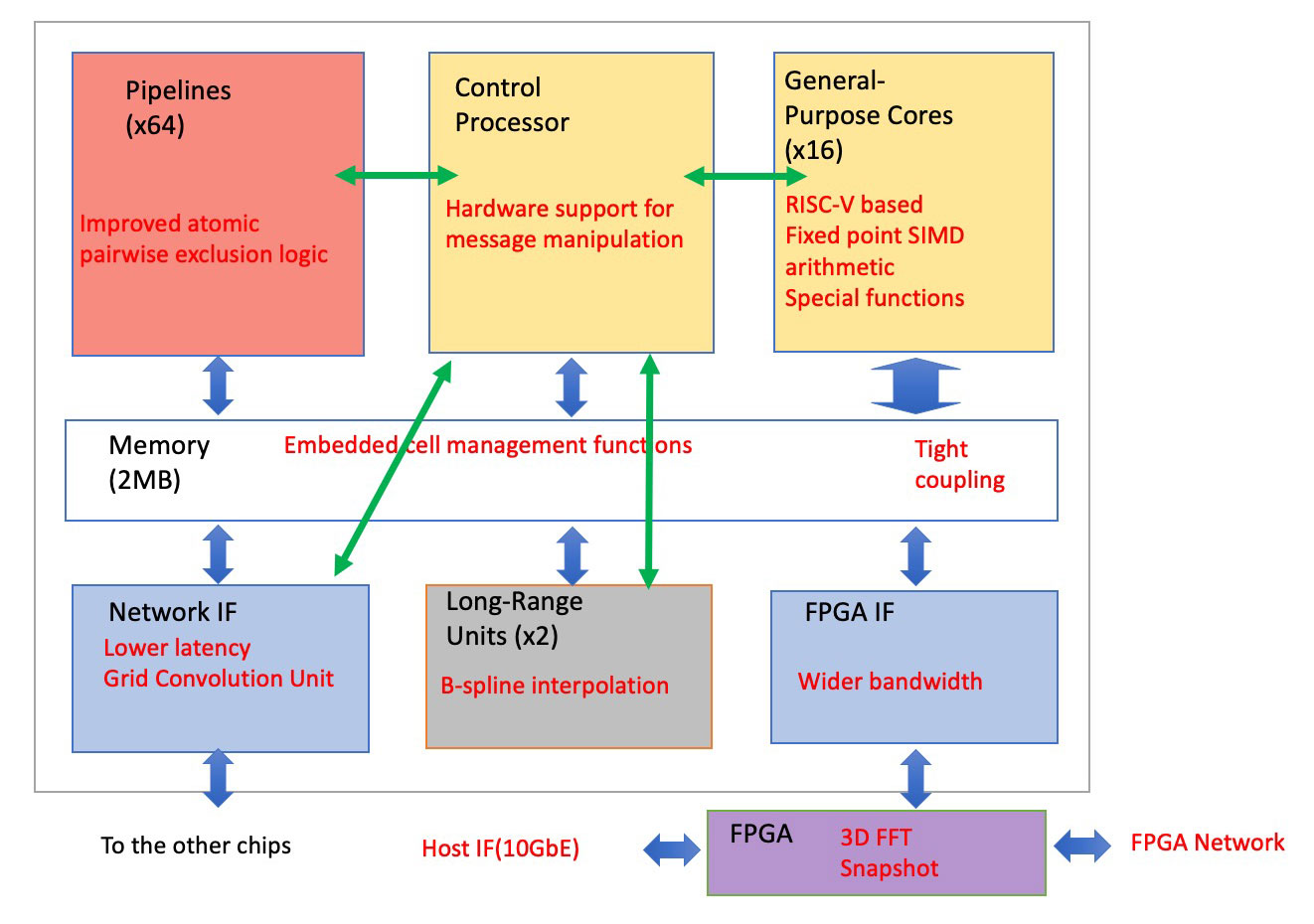
MDGRAPE-4A Board: 8 LSIs

MDGRAPE-4A Full System: 64 Boards

Manufacturer: Tokyo Electron Device, Integran, HOKS, Fujikura
Power: 65kW
Cooling: air-flow
Cost: ~US$6,500,000
Network topology of the full system
3D-torus(8x8x8x) 6Gbps 12ch
FPGA Tree(64-8-1) 10Gbps 4ch
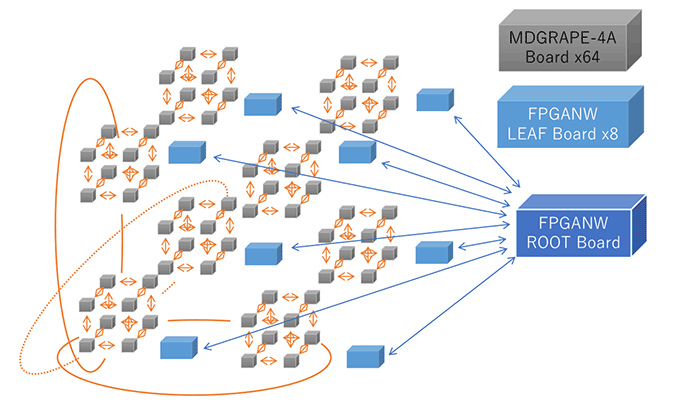
Sequence of parallel MD computation
Achieved performance: less than 200µs to finish 1 MD time step (dt = 2.5fs), more than 1µs/day simulation speed.
左右にスクロールできます
| Computer System | Elapsed time for single step (µs) | Performance (µs/day, dt=2.5fs) |
|---|---|---|
| MDGRAPE-4A | 200 | 1 |
| Commodity Cluster | 1,000 | 0.2 |
| GPU | 2,000 | 0.1 |
| Laptop | 100,000 | 0.002 |
*These are rough order estimate and detailed values are dependent on simulation conditions. Anton, the other special computer developed by D. E. Shaw Research is one or two orders of magnitude ahead of MDGRAPE-4A.
Software specifications
- GROMACS compatible input/output converter
- Velocity-Verlet integrator
- AMBER force field (CHARMM w/o CMAP, OPLS-AA can be converted)
- SETTLE constraint for TIP3P water
- RATTLE constraint for H-bonds
- Temperature control (velocity rescaling)
- Long range electrostatics (TME)
- Pressure control (Monte Carlo barostat)
- Extension for larger complicated systems (>120k atoms, membrane)
- Extension for other various force fields (TIP4P, CMAP)
Algorithms for electrostatic interactions
- ◆ Tensor-structured multilevel Ewald summation method (TME)[3]
- TME can be considered as a combination of the smooth particle mesh Ewald method (SPME)[4] and the B-spline multilevel summation method (B-spline MSM)[5] with approximations by Gaussian functions. For MDGRAPE-4A system, the precision is comparable to SPME with 6 order B-spline (5 degree piecewise polynomial) interpolation.
- ◆ Zero-Multipole Summation Method (ZMM)[6]
- Cutoff method utilizing electrostatic neutralization principle. Long-range electrostatic is not calculated.
左右にスクロールできます
| Coulomb | Performance (µs/day) |
|---|---|
| ZMM(order=2) | 1.25 |
| TME (6 order B-spline, 32 x 32 x 32 grid) | 1.12 |
Target system to measure the full system performance: 100,000 atoms in total in ~ 10nm cubic box. The system is divided into 4096 cells, 8 cells per node.
Against COVID-19
Simulated binding process of a HIV inhibitor (nelfinavir) to SARS-CoV-2 Mpro
One of the applications of long-term MD simulations is drug discovery study. The purposes of the simulation are to explore the change of site pocket shape, to find the binding pathway and to estimate the affinity of a ligand to the site.(see [2] in more detail)
References
- 1.Ohmura, I., et al., MDGRAPE-4: a special-purpose computer system for molecular dynamics simulations, Phil. Trans. R. Soc. A, 372:20130387, 2014.
- 2.Komatsu, T. S. et al., Drug binding dynamics of the dimeric SARS-CoV-2 main protease determined by molecular dynamics simulation, Sci. Rep. 10, 16986, 2020.
- 3.Y. M. Koyama et al., in preparation (see also Supplementary information 'Z' in [2])
- 4.Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. (1995) doi:10.1063/1.470117.
- 5.Hardy, D. J., et al., Multilevel summation with B-spline interpolation for pairwise interactions in molecular dynamics simulations, J. Chem. Phys.,144:114112, 2016.
- 6.Sakuraba, S. et al., Performance evaluation of the zero-multipole summation method in modern molecular dynamics software, J. Comp. Chem. 39 (20), 2018.
Acknowledgments
Some of this work is conducted as a part of “Construction of Drug Discovery Informatics System” supported by Japan Agency for Medical Research and Development (AMED). This work is supported by the Japan Society for the Promotion of Science (JSPS). We wish to thank Taisho Pharmaceutical Co., Ltd. for the collaboration to evaluate the design of the LSI.
LINK
Contact address: mdgrape-contact[at]ml.riken.jp (Please replace [at] with @.)