



Team Director
Teruki Honma
Ph.D.
Laboratory for Structure-Based Molecular Design
[Affiliation has changed to RIKEN Center for Integrated Medical Sciences (IMS) as of April 2025]
Location Yokohama
E-mailhonma.teruki@riken.jp
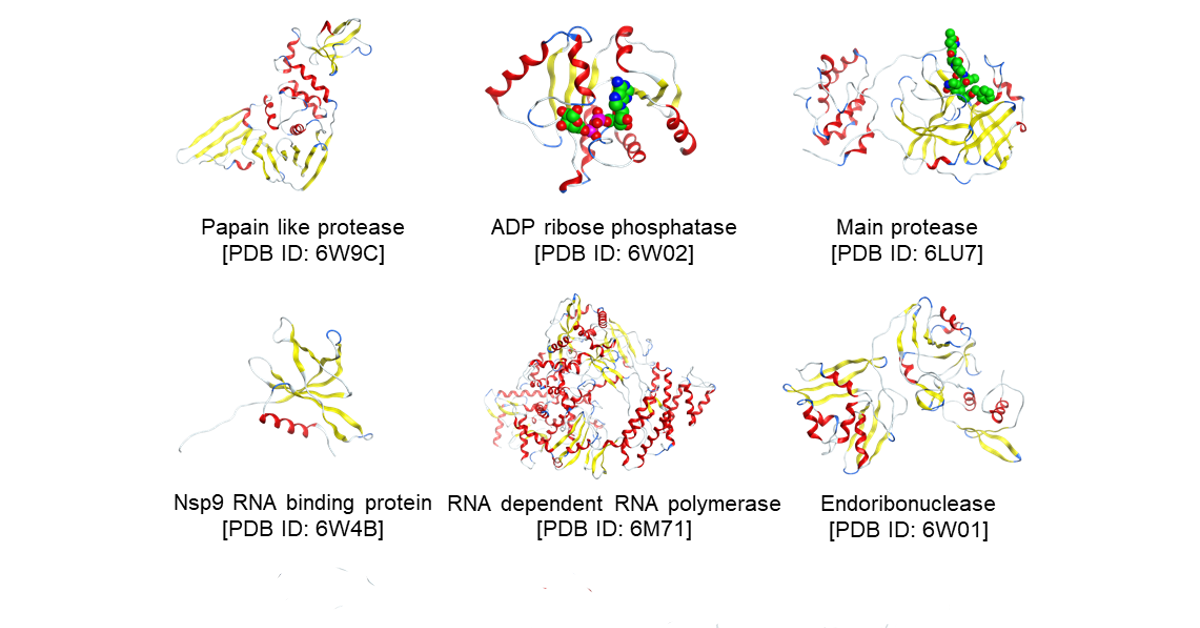
Through the long history of small molecule drug discovery, conventional “druggable” targets had been already investigated and many drugs were marketed. On the other hand, in addition to refractory cancer, Alzheimer's disease, genetic diseases, the risk of new infections is increasing, so there are strong needs for innovative new drugs that have never existed. The team develops new technologies for in silico design and drug discovery artificial intelligence (AI) by combining simulation such as molecular dynamics and quantum chemical calculation (FMO method) and informatics technology represented by AI. The developed technologies are applied to in silico screening for drug discovery targets with high difficulty by conventional. Using the hits obtained by the screening, simultaneous optimization of multiple items necessary for medicines such as potency, pharmacokinetics, toxicity etc. is carried out. In addition, we develop and operate the world's first quantum chemistry calculation database (FMO IFIE database) of proteins.
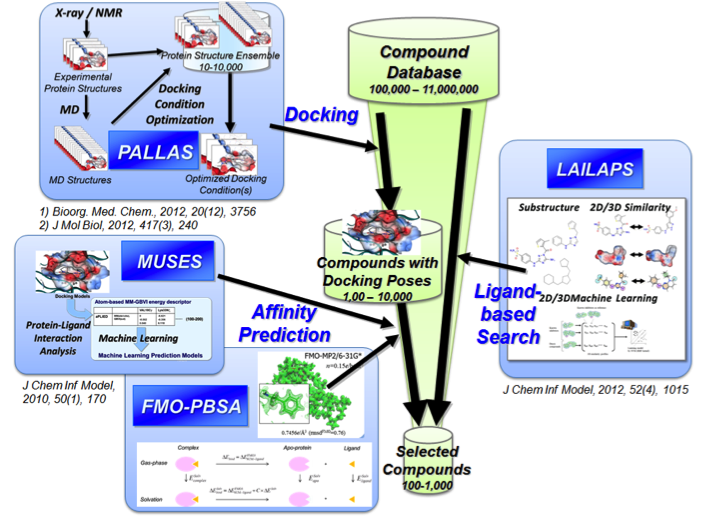
PALLAS: a system for docking condition optimization
MUSES: affinity prediction using interaction descriptors
FMO-PBSA: affinity prediction based on QM and solvent effects
LAILAPS: a system for muti-directional ligand searching
Research Theme
- Development of new technologies for in silico drug discovery combining simulation and informatics
- Application of in silico drug discovery technologies to drug discovery targets
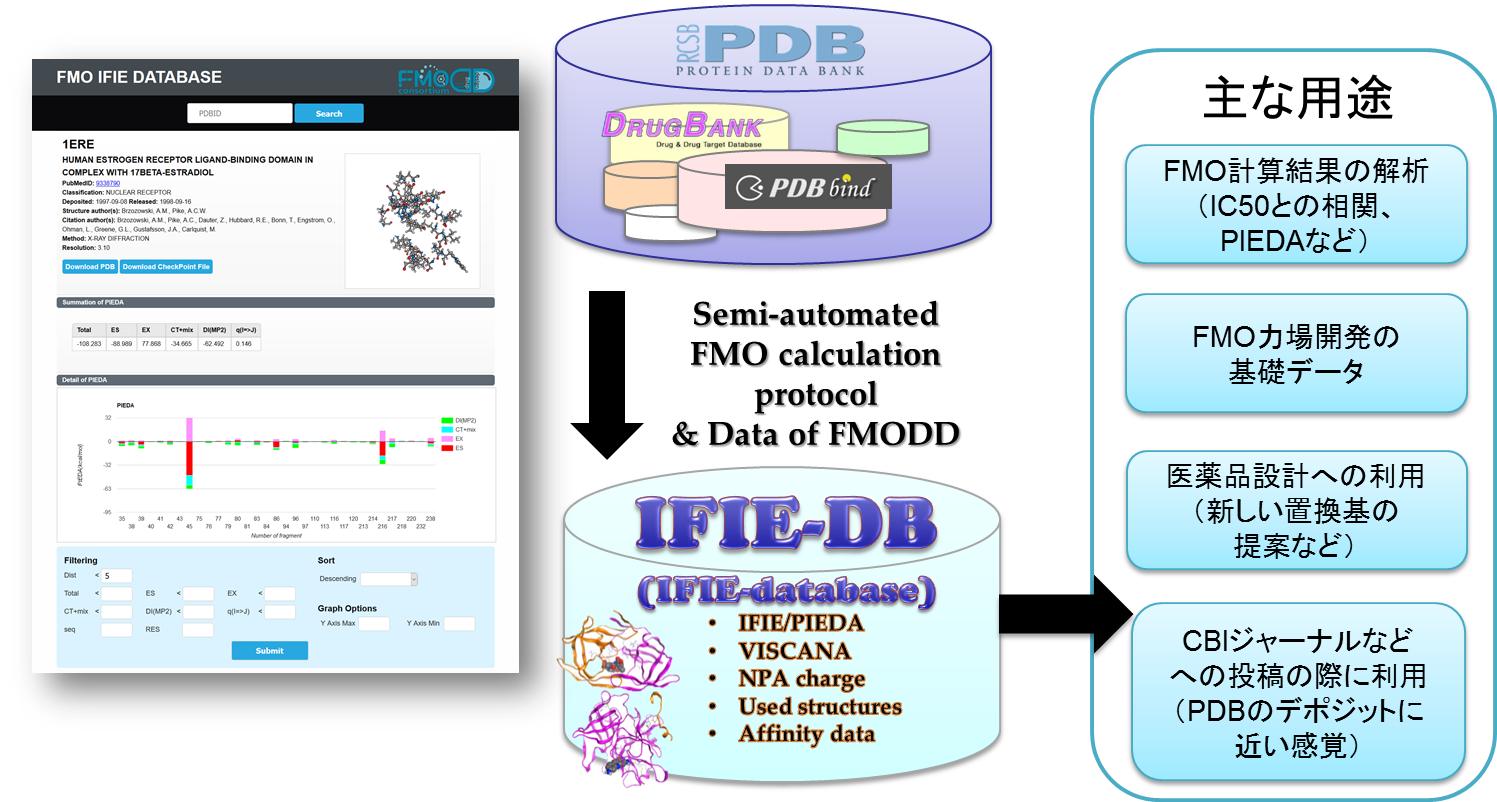
- Construction and publication of FMO IFIE database
Selected Publications
Komura H, Watanabe R, Kawashima H, et al.
A public-private partnership to enrich the development of in silico predictive models for pharmacokinetic and cardiotoxic properties.
Drug discovery today
26(5), 1275-1283 (2021)
doi: 10.1016/j.drudis.2021.01.024
Sato T, Hashimoto N, Honma T.
Bioisostere Identification by Determining the Amino Acid Binding Preferences of Common Chemical Fragments.
Journal of Chemical Information and Modeling
57(12), 2938-2947 (2017)
doi: 10.1021/acs.jcim.7b00092
Watanabe C, Watanabe H, Fukuzawa K, et al.
Theoretical Analysis of Activity Cliffs among Benzofuranone Class Pim1 Inhibitors Using the Fragment Molecular Orbital Method with Molecular Mechanics Poisson-Boltzmann Surface Area (FMO+MM-PBSA) Approach.
Journal of Chemical Information and Modeling
57(12), 2996-3010 (2017)
doi: 10.1021/acs.jcim.7b00110
Okada-Iwabu M, Yamauchi T, Iwabu M, et al.
A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity.
Nature
503, 493-499 (2013)
doi: 10.1038/nature12656
Saito Y, Yuki H, Kuratani M, et al.
A pyrrolo-pyrimidine derivative targets human primary AML stem cells in Vivo..
Science Translational Medicine
5(181), 181ra152 (2013)
doi: 10.1126/scitranslmed.3004387
Shiba T, Kido Y, Sakamoto K, et al.
Structure of the trypanosome cyanide-insensitive alternative oxidase.
Proceedings of the National Academy of Sciences of the United States of America
110(12), 4580-5 (2013)
doi: 10.1073/pnas.1218386110
Takaya D, Sato T, Yuki H, et al.
Prediction of Ligand-Induced Structural Polymorphism of Receptor Interaction Sites Using Machine Learning.
Journal of Chemical Information and Modeling
53(3), 704-716 (2013)
doi: 10.1021/ci300458g
Sato T, Watanabe H, Tsuganezawa K, et al.
Identification of novel drug-resistant EGFR mutant inhibitors by in silico screening using comprehensive assessments of protein structures.
Bioorganic & Medicinal Chemistry
20(12), 3756-67 (2012)
doi: 10.1016/j.bmc.2012.04.042
Yuki H, Honma T, Hata M, Hoshino T.
Prediction of sites of metabolism in a substrate molecule, instanced by carbamazepine oxidation by CYP3A4.
Bioorganic & Medicinal Chemistry
20(2), 775-83 (2011)
doi: 10.1016/j.bmc.2011.12.004
Sato T, Honma T, Yokoyama S.
Combining Machine Learning and Pharmacophore-based Interaction Fingerprint for in silico Screening.
Journal of Chemical Information and Modeling
50(1), 170-85 (2010)
doi: 10.1021/ci900382e
Sato T, Matsuo Y, Honma T, Yokoyama S.
In silico functional profiling of small molecules and its applications.
Journal of Medicinal Chemistry
51(24), 7705-16 (2008)
doi: 10.1021/jm800504q